
La phénylcétonurie est une maladie génétique rare qui touche en moyenne 1 nouveau-né sur 17000 en France. Elle est dépistée systématiquement et nécessite un suivi médical régulier tout au long de la vie.
Document 1 : cause de la phénylcétonurie
La phénylalanine est une molécule issue de l’alimentation. Elle est transformée par l’organisme en une autre molécule, la tyrosine, grâce à l’activité d’une enzyme nommée PAH :

Chez les personnes atteintes de phénylcétonurie, l’enzyme PAH n’est pas fonctionnelle :
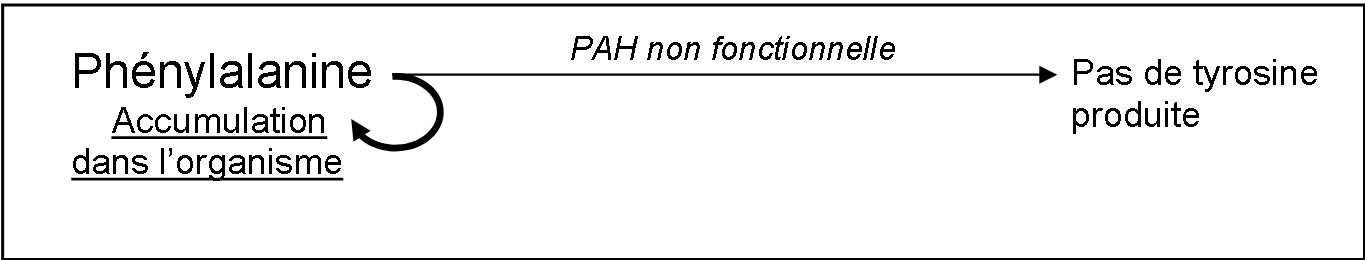
Un excès de phénylalanine lors du développement du nouveau-né cause des dommages cérébraux chez l’enfant.
Document 2 : diagnostic de la phénylcétonurie en fonction de la concentration sanguine en phénylalanine
Depuis 1972, le dépistage de la phénylcétonurie est systématique : il est réalisé dès la naissance chez tous les nouveau-nés grâce à un examen sanguin.
Document 2a : résultats des tests de dépistage et traitements conseillés dès la naissance
|
Taux sanguin de phénylalanine (PHE) en mg.dL-1 |
PHE < 2 |
2 < PHE <10 |
PHE > 10 |
|
Dépistage |
Négatif |
Positif |
|
|
Traitement |
Aucun traitement |
Pas de régime alimentaire, mais suivi médical |
Régime alimentaire pauvre en phénylalanine et riche en tyrosine Suivi médical |
Document 2b : analyse sanguine de 3 nouveau-nés dont le test de dépistage est positif
|
Patient 1 |
Patient 2 |
Patient 3 |
|
| Taux sanguin de phénylalanine (en mg.dL-1) |
4 |
19 |
8 |
Question 1a : (5 points)
À partir du document 2, citer le traitement que le patient 2 devra suivre. Justifier votre réponse avec des valeurs chiffrées.
Question 1b : (6 points)
À l’aide des documents 1 et 2, indiquer sur votre copie, les deux propositions qui conviennent à la situation suivante.
Un patient dont le taux sanguin en phénylalanine est supérieur à 10 mg.dL-1 doit respecter une alimentation :
a. pauvre en phénylalanine pour éviter l’accumulation de tyrosine toxique pour le cerveau.
b. pauvre en phénylalanine pour éviter l’accumulation de phénylalanine et éviter le développement des dommages cérébraux.
c. riche en tyrosine, car l’enzyme PAH n’est pas fonctionnelle et la phénylalanine ne peut être transformée en tyrosine.
d. riche en tyrosine, car l’enzyme PAH n’est pas fonctionnelle et la tyrosine ne peut être transformée en phénylalanine.
Document 3 : étude d’un cas rare
Un médecin étudie un cas très particulier, celui d’un adulte (patient A) diagnostiqué positif tardivement à l’âge de 10 ans.
Document 3a : quelques données sur le patient A
-
-
Patient A
Diagnostic atteint de phénylcétonurie
Suivi médical irrégulier
Régime alimentaire nécessaire commencé tardivement
Symptômes de la phénylcétonurie développés
Peau, cheveux, yeux très clairs
-
Document 3b : rôle de la mélanine sur la coloration de la peau, des cheveux et des yeux
La mélanine est une molécule produite par l’organisme à partir de la tyrosine. Plus la concentration de mélanine est importante, plus la coloration de la peau, des cheveux et des yeux sera importante (plus foncée).
Question 2 : (7 points)
A l’aide des documents 1, 2 et 3, expliquer le lien entre la phénylcétonurie du patient A et l’aspect très clair de sa peau, ses cheveux et ses yeux.
Document 4 : phénylcétonurie et suivis médicaux
• Durant toute sa vie, un individu atteint de phénylcétonurie doit surveiller régulièrement son taux de phénylalanine pour éviter toute complication en particulier neurologique et comportementale. Il peut faire des prélèvements à domicile (deux taches de sang sur un carton buvard envoyé en laboratoire).
• Chez une femme enceinte atteinte de phénylcétonurie, il existe un risque pour le développement du futur bébé. Un régime alimentaire strict et un contrôle du taux de phénylalanine tout au long de la grossesse sont donc nécessaires pour éviter la survenue de taux élevés ou trop bas pouvant altérer la croissance du fœtus.
Question 3 : (7 points)
Depuis environ 60 ans, des avancées médicales permettent d’améliorer les conditions de vie des patients atteints de phénylcétonurie. A partir de l’ensemble des documents, expliquer comment les symptômes graves de cette maladie peuvent aujourd’hui être évités.
Trois idées sont attendues.
Version imprimable : brevet site phenylcetonurie